Showing posts with label Bioinformatics. Show all posts
Showing posts with label Bioinformatics. Show all posts
Sunday, 10 November 2019
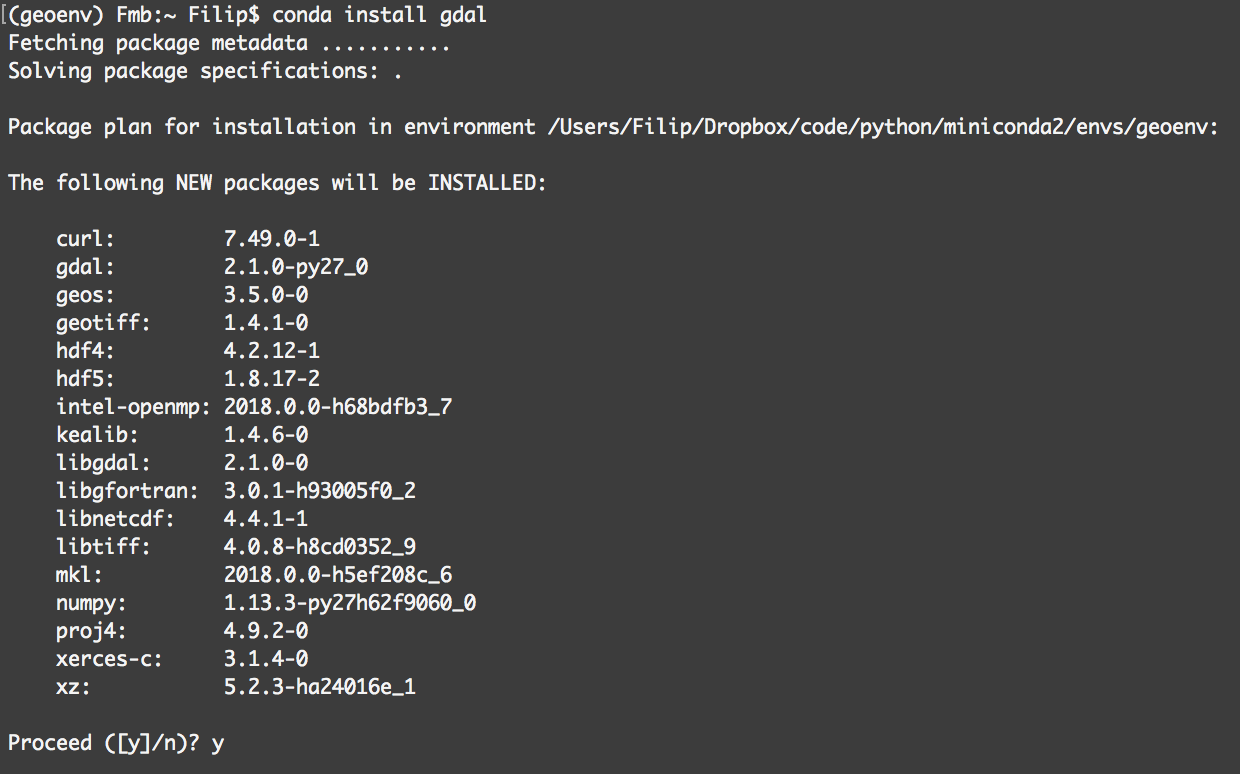
10 minutes guide to Bioconda
Bioinformatics is complicated, what with its arcane command-line interface, complex workflows, and massive datasets. For new bioinformaticians, installing the software can present a problem.
But the good news is that the Bioinformatics community has already a solution to this problem: BioConda + BioContainers.
Wednesday, 6 November 2019
List of major preprints servers - where to go
The most well-known preprint server is probably arXiv (pronounced like ‘archive’). It started as a server for preprints in physics and has since expanded out to various subjects, including mathematics, computer science, and economics. The arXiv server is now run by the Cornell University Library and contains 1.37 million preprints so far.

The Open Science Framework provides an open-source framework to help researchers and institutions set up their own preprint servers. One such example is SocArXiv for the Social Sciences. On their website, you can browse more than 2 million preprints, including preprints on arXiv, and many of them have their own preprint digital object identifier (DOI). In cases where the preprint has now been published it also links to the publication’s DOI.
Cold Spring Harbor Laboratory set up bioaRxiv, a preprint server for Biology in 2013 to complement arXiv. The bioaRxiv server has a direct transfer service to several journals such as Science and PNAS and a bit over 60% of papers in bioaRxiv end up published in peer-reviewed journals.
In more recent years a lot of new servers have popped up covering almost every field including the social sciences, arts, and humanities fields. Here’s a quick overview of some of the rest:
arXiv -> Mathematics, Computer science, and economics, Physics
EngrXiv - Engineering
ChemRxiv - Chemical sciences
PsyArXiv - Psychological sciences
SportaRxiv - Sport and exercise science
PaleoarXiv - Paleontology
LawArXiv - Law
AgriXiv - Agricultural sciences
NutriXiv - Nutritional sciences
MarXiv - Ocean and marine-climate sciences
EarthArXiv - Earth sciences
Preprints.org - Arts & Humanities, Behavioral Sciences, Biology, Chemistry, Earth Sciences, Engineering, Life Sciences, Materials Science, Mathematics & Computer Science, Medicine &, Pharmacology, Physical Sciences, Social Sciences
Thursday, 9 June 2016
How to estimate and compute the isoelectric point of peptides and proteins?
By +Yasset Perez-Riverol and +Enrique Audain :
Isoelectric point (pI) can be defined as the point of singularity in a titration curve, corresponding to the solution pH value at which the net overall surface charge is equal to zero. Currently, there are available different modern analytical biochemistry and proteomics methods depend on the isoelectric point as a principal feature for protein and peptide characterization. Peptide/Protein fractionation according to their pI is widely used in current proteomics sample preparation procedures previous to the LC-MS/MS analysis. The experimental pI records generated by pI-based fractionation procedures are a valuable information to validate the confidence of the identifications, to remove false positive and and could be used to re-compute peptide/protein posterior error probabilities in MS-based proteomics experiments.
Isoelectric point (pI) can be defined as the point of singularity in a titration curve, corresponding to the solution pH value at which the net overall surface charge is equal to zero. Currently, there are available different modern analytical biochemistry and proteomics methods depend on the isoelectric point as a principal feature for protein and peptide characterization. Peptide/Protein fractionation according to their pI is widely used in current proteomics sample preparation procedures previous to the LC-MS/MS analysis. The experimental pI records generated by pI-based fractionation procedures are a valuable information to validate the confidence of the identifications, to remove false positive and and could be used to re-compute peptide/protein posterior error probabilities in MS-based proteomics experiments.
Theses approaches require an accurate theoretical prediction of pI. Even thought
there are several tools/methods to predict the isoelectric point, it remains
hard to define beforehand what methods perform well on a specific dataset.
We believe that the best way to compute the isoelectric point (pI) is to have a complete package with most of the algorithms and methods in the state of the art that can do the job for you [2]. We recently developed an R package (pIR) to
compute isoelectric point using long-standing and novels pI methods that can be
grouped in three main categories : a) iterative, b) Bjellvist-based methods and c) machine learning methods. In addition, pIR also offers a statistical
and graphical framework to evaluate the performance of each method and its
capability to “detect” outliers (those peptides/protein with theoretical pI
biased from experimental value) in a high-throughput environment.
First lets install the package:
First, we need to install
devtools:install.packages("devtools")
library(devtools)
Then we just call:
install_github("ypriverol/pIR")
library(pIR)Sunday, 3 April 2016
Wednesday, 21 October 2015
Installing MESOS in your Mac
1- Homebrew is an open source package management system for the Mac that simplifies installation of packages from source.
ruby -e "$(curl -fsSL https://raw.github.com/Homebrew/homebrew/go/install)"
2- Once you have Homebrew installed, you can install Mesos on your laptop with these two commands:
brew update
brew install mesos
You will need to wait while the most current, stable version of Mesos is downloaded, compiled, and installed on your machine. Homebrew will let you know when it’s finished by displaying a beer emoji in your terminal and a message like the following:
/usr/local/Cellar/mesos/0.19.0: 83 files, 24M, built in 17.4 minutes
Start Your Mesos Cluster
3- Running Mesos in your machine: Now that you have Mesos installed on your laptop, it’s easy to start your Mesos cluster. To see Mesos in action, spin up an in-memory master with the following command:
/usr/local/sbin/mesos-master --registry=in_memory --ip=127.0.0.1
A Mesos cluster needs at least one Mesos Master to coordinate and dispatch tasks onto Mesos Slaves. When experimenting on your laptop, a single master is all you need. Once your Mesos Master has started, you can visit its management console: http://localhost:5050

Since a Mesos Master needs slaves onto which it will dispatch jobs, you might also want to run some of those. Mesos Slaves can be started by running the following command for each slave you wish to launch:
sudo /usr/local/sbin/mesos-slave --master=127.0.0.1:5050
ruby -e "$(curl -fsSL https://raw.github.com/Homebrew/homebrew/go/install)"
2- Once you have Homebrew installed, you can install Mesos on your laptop with these two commands:
brew update
brew install mesos
You will need to wait while the most current, stable version of Mesos is downloaded, compiled, and installed on your machine. Homebrew will let you know when it’s finished by displaying a beer emoji in your terminal and a message like the following:
/usr/local/Cellar/mesos/0.19.0: 83 files, 24M, built in 17.4 minutes
Start Your Mesos Cluster
3- Running Mesos in your machine: Now that you have Mesos installed on your laptop, it’s easy to start your Mesos cluster. To see Mesos in action, spin up an in-memory master with the following command:
/usr/local/sbin/mesos-master --registry=in_memory --ip=127.0.0.1
A Mesos cluster needs at least one Mesos Master to coordinate and dispatch tasks onto Mesos Slaves. When experimenting on your laptop, a single master is all you need. Once your Mesos Master has started, you can visit its management console: http://localhost:5050

Since a Mesos Master needs slaves onto which it will dispatch jobs, you might also want to run some of those. Mesos Slaves can be started by running the following command for each slave you wish to launch:
sudo /usr/local/sbin/mesos-slave --master=127.0.0.1:5050
Wednesday, 30 September 2015
First Scrum Board

Team members update the task board continuously each sprint; if someone thinks of a new task (“test a new machine learning algorithm”), she writes a new card and puts it on the wall. Either during or before the daily scrum, estimates are changed (up or down), and cards are moved around the board.
Each row on the Scrum board is a user story, which is the unit of work we encourage teams to use for their product backlog.
During the sprint planning meeting, the team selects the product backlog items they can complete during the next Spring. Each product backlog item is turned into multiple sprint backlog items. Each of these is represented by one task card that is placed on the Scrumboard.
- Story (User Story): The story description (“As a user we want to…”) shown on that row.
- Ongoing: Any card being worked on goes here. The programmer who chooses to work on it moves it over when she's ready to start the task. Often, this happens during the daily scrum when someone says, “I'm going to work on the boojum today.”
- Testing: A lot of tasks have corresponding test task cards. So, if there's a “Code the boojum class” card, there is likely one or more task cards related to testing: “Test the boojum”, “Write FitNesse tests for the boojum,” “Write FitNesse fixture for the boojum,”
- Done: Cards pile up over here when they're done. They're removed at the end of the sprint. Sometimes we remove some or all during a sprint if there are a lot of cards.
Optionally, depending on the team, the culture, the project and other considerations:
- Notes: Just a place to jot a note or two.
- Tests Specified: We like to do “Story Test-Driven Development,” or “Acceptance Test-Driven Development,” which means the tests are written before the story is coded. Many teams find that it helps to have acceptance tests identified before coding begins on a particular story. This column just contains a checkmark to indicate the tests are specified.
Saturday, 29 August 2015
DIA-Umpire Pipeline Using BioDocker containers.
Sunday, 31 May 2015
I love technical notes and short manuscripts

One of my first papers in 2012 (here), was related with support vector (SVM) machines. It was a simple algorithm, that improved the method to compute the isoelectric point of peptides using SVM. The first time I presented the results to my colleagues, one of them ask me: "are you planning to publish this?". One of the senior co-authors said, "we can write a big research manuscript, explaining other algorithms, compare them, use other datasets, etc". Another said (computer scientist), "we can explore other features from peptides including topological indexes.. and write a full research manuscript about.."....
"I was very clear from the very beginning, We will write a Technical Note or Letter. "
Sunday, 10 May 2015
A Trans-Proteomic Pipeline (TPP) Docker container
By +Felipe Leprevost & +Yasset Perez-Riverol
In my initial post in this blog, I will teach you how to use a Docker container with the Trans-Proteomic Pipeline software installation.
Docker is a great new technology that allows us to create GNU/Linux containers with specific software inside. All kinds of software can be "containerized", including ones that rely on graphical user interfaces.
The whole idea of using a Docker container is built on having a software that is isolated from the host OS and can interact with the outside world. GNU/Linux containers, like Docker, are very useful even in the scientific world where bioinformatics applications are used every day.
Using Docker with bioinformatics software helps to solve some issues we face, like reproducibility, for example. We wrote about this last year [1] . You can also check for more containers with bioinformatics applications in the BioDocker webpage.
Here I am going to describe to you how to install and use one of the most powerful software for proteomics data analysis, the Trans-Proteomics Pipeline (TPP).
With a Docker TPP container you can just download it and use it on the command line, the container it self behaves like an executable, so image the possibilities.
Lets begin preparing your environment for Docker. The first thing you have to do is to install some libraries that are essential for the Docker daemon to run properly. If you are running an Ubuntu OS, you can skip this step. If you are on a different OS, like Linux Mint for example you need to follow these steps.
In my initial post in this blog, I will teach you how to use a Docker container with the Trans-Proteomic Pipeline software installation.
Docker is a great new technology that allows us to create GNU/Linux containers with specific software inside. All kinds of software can be "containerized", including ones that rely on graphical user interfaces.
The whole idea of using a Docker container is built on having a software that is isolated from the host OS and can interact with the outside world. GNU/Linux containers, like Docker, are very useful even in the scientific world where bioinformatics applications are used every day.
Using Docker with bioinformatics software helps to solve some issues we face, like reproducibility, for example. We wrote about this last year [1] . You can also check for more containers with bioinformatics applications in the BioDocker webpage.
Here I am going to describe to you how to install and use one of the most powerful software for proteomics data analysis, the Trans-Proteomics Pipeline (TPP).
Unfortunately, if you are a GNU/Linux user (like me) and your job involve MS/MS data analysis (also, like me), you will probably have some harsh time trying to install TPP. Almost all the tutorials available on the Web focus on the Windows users, so novice bioinformaticians or those that are not too versatile with GNU/Linux can have some hard times.
With a Docker TPP container you can just download it and use it on the command line, the container it self behaves like an executable, so image the possibilities.
Lets begin preparing your environment for Docker. The first thing you have to do is to install some libraries that are essential for the Docker daemon to run properly. If you are running an Ubuntu OS, you can skip this step. If you are on a different OS, like Linux Mint for example you need to follow these steps.
Wednesday, 29 October 2014
What is BioHackathon 2014?
In a week BioHackathon 2014 will start (http://www.biohackathon.org/). It will be my first time ins this kind of "meeting". I will give a talk about PRIDE and ProteomeXchange and future developments of both platforms (below the complete list of talks).
But first, a quick introduction of BioHackathon. National Bioscience Database Center (NBDC) and Database Center for Life Science (DBCLS) have been organizing annual BioHackathon since 2008, mainly focusing on standardization (ontologies, controlled vocabularies, metadata) and interoperability of bioinformatics data and web services for improving integration (semantic web, web services, data integration), preservation and utilization of databases in life sciences. This year, we will focus on the standardization and utilization of human genome information with Semantic Web technologies in addition to our previous efforts on semantic interoperability and standardization of bioinformatics data and Web services.
Sunday, 14 September 2014
ProteoWizard: The chosen one in RAW file conversion
 |
| I'm the chosen one. |
File formats (the way
that we use to represent, storage and exchange our data) are fundamentals
piece in bioinformatics, more than that, are one of the milestone of
the Information Era. In some fields the topic is more stable than others, but
the topic is still in the table for most of us. To have a quick idea see the
evolution of general standards in recent years like XML,
JSON and recently YAML.
Tuesday, 26 August 2014
Adding CITATION to your R package

Software is very important in science – but good software takes time and effort that could be used to do other work instead. I believe that it is important to do this work – but to make it worthwhile, people need to get credit for their work, and in academia that means citations. However, it is often very difficult to find out how to cite a piece of software – sometimes it is hidden away somewhere in the manual or on the web-page, but often it requires sending an email to the author asking them how they want it cited. The effort that this requires means that many people don’t bother to cite the software they use, and thus the authors don’t get the credit that they need. We need to change this, so that software – which underlies a huge amount of important scientific work – gets the recognition it deserves.
Wednesday, 20 August 2014
ProteoStats: Computing false discovery rates in proteomics
By Amit K. Yadav (@theoneamit) & Yasset Perez-Riverol (@ypriverol):
Perl is a legacy language thought to be abstruse by many modern programmers. I’m passionate with the idea of not letting die a programming language such as Perl. Even when the language is used less in Computational Proteomics, it is still widely used in Bioinformatics. I’m enthusiastic writing about new open-source libraries in Perl that can be easily used. Two years ago, I wrote a post about InSilicoSpectro and how it can be used to study protein databases like I did in “In silico analysis of accurate proteomics, complemented by selective isolation of peptides”.
Perl is a legacy language thought to be abstruse by many modern programmers. I’m passionate with the idea of not letting die a programming language such as Perl. Even when the language is used less in Computational Proteomics, it is still widely used in Bioinformatics. I’m enthusiastic writing about new open-source libraries in Perl that can be easily used. Two years ago, I wrote a post about InSilicoSpectro and how it can be used to study protein databases like I did in “In silico analysis of accurate proteomics, complemented by selective isolation of peptides”.
Today’s post is about ProteoStats [1], a Perl library for False Discovery Rate (FDR) related calculations in proteomics studies. Some background for non-experts:
One of the central and most widely used approach for shotgun proteomics is the use of database search tools to assign spectra to peptides (called as Peptide Spectrum Matches or PSMs). To evaluate the quality of the assignments, these programs need to calculate/correct for population wise error rates to keep the number of false positives under control. In that sense, the best strategy to control the false positives is the target-decoy approach. Originally proposed by Elias & Gygi in 2007, the so-called classical FDR strategy or formula proposed involved a concatenated target-decoy (TD) database search for FDR estimation. This calculation is either done by the search engine or using scripts (in-house, non-published, not benchmarked, different implementations).
So far, the only library developed to compute FDR at spectra level, peptide level and protein level FDRs is MAYU [2]. But, while MAYU only uses the classical FDR approach, ProteoStats provides options for 5 different strategies for calculating the FDR. The only prerequisite being that you need to search using a separate TD database as proposed by Kall et al (2008) [3]. Also, ProteoStats provides a programming interface that can read the native output from most widely used search tools and provide FDR related statistics. In case of tools not supported, pepXML, which has become a de facto standard output format, can be directly read along with tabular text based formats like TSV and CSV (or any other well-defined separator).
Sunday, 8 June 2014
Thesis: Development of computational methods for analysing proteomic data for genome annotation
Thesis by Markus Brosch in 2009 about Computational proteomics methods for analysing proteomic data for genome annotation.
Notes from Abstract
Notes from Abstract
Proteomic mass spectrometry is a method that enables sequencing of gene product fragments, enabling the validation and refinement of existing gene annotation as well as the detection of novel protein coding regions. However, the application of proteomics data to genome annotation is hindered by the lack of suitable tools and methods to achieve automatic data processing and genome mapping at high accuracy and throughput.
In the first part of this project I evaluate the scoring schemes of “Mascot”, which is a peptide identification software that is routinely used, for low and high mass accuracy data and show these to be not sufficiently accurate. I develop an alternative scoring method that provides more sensitive peptide identification specifically for high accuracy data, while allowing the user to fix the false discovery rate. Building upon this, I utilise the machine learning algorithm “Percolator” to further extend my Mascot scoring scheme with a large set of orthogonal scoring features that assess the quality of a peptide-spectrum match.
To close the gap between high throughput peptide identification and large scale genome annotation analysis I introduce a proteogenomics pipeline. A comprehensive database is the central element of this pipeline, enabling the efficient mapping of known and predicted peptides to their genomic loci, each of which is associated with supplemental annotation information such as gene and transcript identifiers.
In the last part of my project the pipeline is applied to a large mouse MS dataset. I show the value and the level of coverage that can be achieved for validating genes and gene structures, while also highlighting the limitations of this technique. Moreover, I show where peptide identifications facilitated the correction of existing annotation, such as re-defining the translated regions or splice boundaries.
Moreover, I propose a set of novel genes that are identified by the MS analysis pipeline with high confidence, but largely lack transcriptional or conservational evidence.
Java Optimization Tips (Memory, CPU Time and Code)
There are several common optimization techniques that apply regardless
of the language being used. Some of these techniques,
such as global register allocation, are
sophisticated strategies to allocate machine resources (for example, CPU
registers)
and don't apply to Java bytecodes. We'll
focus on the techniques that basically involve restructuring code and
substituting
equivalent operations within a method.
EntrySet vs KeySet
-----------------------------------------
More efficient:
for (Map.Entry entry : map.entrySet()) {
Object key = entry.getKey();
Object value = entry.getValue();
}
than:
for (Object key : map.keySet()) {
Object value = map.get(key);
}
Avoid to create threads without run methods
------------------------------------
Usage Example:
public class Test
{
public void method() throws Exception
{
new Thread().start(); //VIOLATION
}
}
Should be written as:
public class Test
{
public void method(Runnable r) throws Exception
{
new Thread(r).start(); //FIXED
}
}
Initialise the ArrayList if you know in advance the size
--------------------------------------------
For example, use this code if you expect your ArrayList to store around 1000 objects:
List str = new ArrayList(1000)
Use ternary operators
----------------------------------------
class Use_ternary_operator_correction
{
public boolean test(String value)
{
if(value.equals("AppPerfect")) // VIOLATION
{
return true;
}
else
{
return false;
}
}
}
Should be written as:
class Use_ternary_operator_correction
{
public boolean test(String value)
{
return value.equals("AppPerfect"); // CORRECTION
}
}
Always declare constant fields Static
public class Always_declare_constant_field_static_violation
{
final int MAX = 1000; // VIOLATION
final String NAME = "Noname"; // VIOLATION
}
Should be written as:
public class Always_declare_constant_field_static_correction
{
static final int MAX = 1000; // CORRECTION
static final String NAME = "Noname"; // VIOLATION
}
Wednesday, 22 January 2014
What is a bioinformatician
By Anthony Fejes originally posted in blog.fejes.ca
I’ve been participating in an interesting conversation on linkedin, which has re-opened the age old question of what is a bioinformatician, which was inspired by a conversation on twitter, that was later blogged. Hopefully I’ve gotten that chain down correctly.
I’ve been participating in an interesting conversation on linkedin, which has re-opened the age old question of what is a bioinformatician, which was inspired by a conversation on twitter, that was later blogged. Hopefully I’ve gotten that chain down correctly.
In any case, it appears that there are two competing schools of
thought. One is that bioinformatician is a distinct entity, and the
other is that it’s a vague term that embraces anyone and anything that
has to do with either biology or computer science. Frankly, I feel the
second definition is a waste of a perfectly good word, despite being a
commonly accepted method.
Wednesday, 8 January 2014
Are you a Computational Biologist or Bioinformaticist or Bioinformatician?
A recent discussion was provoked by @attilacsordas on twitter January 8 regarding what is the choice term for referring to those researchers working on Bioinformatics and Computational Biology fields.
Are bioinformaticians and bioinformatists alike? :)
— attilacsordas (@attilacsordas) January 8, 2014
This debate is older than people may think and it looks like an insignificant topic, but when you are writing your CV or your internet profile, or you’re looking for a new job, you will need a professional title, and that is really important. If you also look the volume of discussion and opinions about this topic on internet you will realize that the community have different points of view. I've use some of my spare time to read in detail different opinions about it, and also collect some those opinions and articles. Let’s see what common terms are used nowadays for these researchers:
Bioinformaticist, Bioinformatician, Computational Biologist, Digital biologist, bioinformatics analyst
Bioinformaticist, Bioinformatician, Computational Biologist, Digital biologist, bioinformatics analyst
My Formula as a Bioinformatician
Every day, I enjoy reading about bioinformatics in blogs, linkedin, and twitter; away from my daily reading of manuscripts journals. I strongly think that the future of publications/science will be closer & closer to the open access style and this emergent way to publish your ideas faster/brief in your own space. Some of my old co-workers don't understand this way to get in touch with science using informal environments rather than arbitrary/supervised spaces; I just said to them, we make the future, not the past. Reading the popular post “A guide for the lonely bioinformatician”, I was thinking about the last three years and how I have been built my own formula to survive as a lonely bioinformatician in a small country, with a lousy internet connection and without a bioinformatics environment.
All the bioinformaticians that I met during these three years can be categorized in three major groups considering their original background:
2) Physicist, Mathematicians, Computer Scientist, Software Engineers, Software
Developers
3) Philosophers, *
As an embryonic and growing field the diversity is huge, then it is quite complex to express all the data behavior in one model or a formula. Here I will summarize some of the variables of my formula, extremely correlated with the original post suggestions:
Friday, 1 November 2013
Integrating the Biological Universe

Integrating biological data is perhaps one of the most daunting tasks any bioinformatician has to face. From a cursory look, it is easy to see two major obstacles standing in the way: (i) the sheer amount of existing data, and (ii) the staggering variety of resources and data types used by the different groups working in the field (reviewed at [1]). In fact, the topic of data integration has a long-standing history in computational biology and bioinformatics. A comprehensive picture of this problem can be found in recent papers [2], but this short comment will serve to illustrate some of the hurdles of data integration and as a not-so-shameless plug for our contribution towards a solution.
"Reflecting the data-driven nature of modern biology, databases have grown considerably both in size and number during the last decade. The exact number of databases is difficult to ascertain. While not exhaustive, the 2011 Nucleic Acids Research (NAR) online database collection lists 1330 published biodatabases (1), and estimates derived from the ELIXIR database provider survey suggest an approximate annual growth rate of ∼12% (2). Globally, the numbers are likely to be significantly higher than those mentioned in the online collection, not least because many are unpublished, or not published in the NAR database issue." [1]
"Reflecting the data-driven nature of modern biology, databases have grown considerably both in size and number during the last decade. The exact number of databases is difficult to ascertain. While not exhaustive, the 2011 Nucleic Acids Research (NAR) online database collection lists 1330 published biodatabases (1), and estimates derived from the ELIXIR database provider survey suggest an approximate annual growth rate of ∼12% (2). Globally, the numbers are likely to be significantly higher than those mentioned in the online collection, not least because many are unpublished, or not published in the NAR database issue." [1]
Some basic concepts
Traditionally, biological database integration efforts come in three main flavors:
- Federated: Sometimes termed portal, navigational or link integration, it is based on the use of hyperlinks to join data from disparate sources; early examples include SRS and Entrez. Using the federated approach, it is relatively easy to provide current, up-to-date information, but maintaining the hyperlinks requires considerable effort.
- Mediated or View Integration: Provides a unified query interface and collects the results from various data sources (BioMart).
- Warehouse: In this approach different data sources are stored in one place; examples include BioWarehouse and JBioWH. While it provides faster querying over joined datasets, it also requires extra care to maintain the underlying databases completely updated.
Monday, 28 October 2013
One step ahead in Bioinformatics using Package Repositories
About a year ago I published a post about in-house tools in research and how using this type of software may end up undermining the quality of a manuscript and the reproducibility of its results. While I can certainly relate to someone reluctant to release nasty code (i.e. not commented, not well-tested, not documented), I still think we must provide (as supporting information) all “in-house” tools that have been used to reach a result we intend to publish. This applies especially to manuscripts dealing with software packages, tools, etc. I am willing to cut some slack to journals such as Analytical Chemistry or Molecular Cell Proteomics, whose editorial staffs are –and rightly so- more concerned about quality issues involving raw data and experimental reproducibility, but in instances like Bioinformatics, BMC Bioinformatics, several members of the Nature family and others at the forefront of bioinformatics, methinks we should hold them to a higher standard. Some of these journals would greatly benefit from implementing a review system from the point of view of Software Production, moving bioinformatics and science in general one step forward in terms of reproducibility and software reusability. What do you think would happen if the following were checked during peer reviewing?
Subscribe to:
Posts (Atom)